Overview
Copy number variation (CNV) is a genomic alteration phenomenon that can lead to copy number abnormalities in one or more genes involved in the process of evolutionary adaptation, genomic disease, and disease progression. Structural genomic rearrangements (e.g., duplications, deletions, translocations, and inversions) may result in CNVs. detection of CNVs is challenging due to their different sizes compared to single nucleotide polymorphisms (SNPs). In addition, CNVs play a role in causing various diseases. Diseases that have been identified to be associated with copy number variation include schizophrenia, type I diabetes, autism, cardiovascular disease, congenital anomalies, and neurodegenerative diseases. CNVs have thus become a major target for researchers focused on identifying putative sources of disease in individual genomes.
Currently, SNP arrays, short-read, and long-read-length genome sequencing are genome-wide high-throughput technologies that can be used to analyze CNVs in an individual's genome. Each of these technologies has its own limitations and advantages. Among them, long read-length sequencing by PacBio and Oxford Nanopore platforms has been widely welcomed in CNV detection due to its unique advantages.
Major Approaches for CNV Detection: from Cytogenetics to the Latest Sequencing Technologies
Cytogenetic Methods
The first CNV detection method is based on the analysis of the mid-cell plate. At mid-cell division, condensed chromosomes line up along the cell equator, facilitating easy visualization of chromosome structure by light microscopy. Methods of chromosome staining include Giemsa staining, C-banding of mitotic or T-banding staining of telomere regions, chromosome-specific sequence along the length (R, Q, and G bands) techniques, fluorescence in situ hybridization (FISH) techniques, and others. The resolution of this method is low and the average size of CNVs detected is 5-10 Mb.
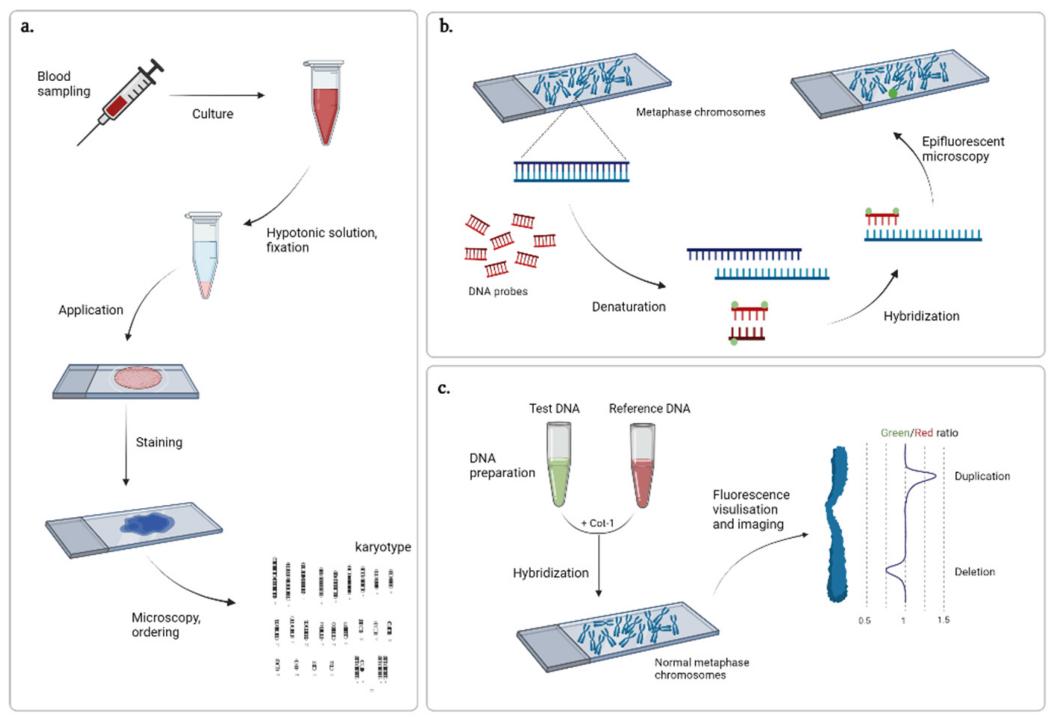
Cytogenetic techniques. (Gordeeva V et al., 2022)
SNP Arrays
SNP arrays leverage probe intensity values in juxtaposition to a predetermined reference to extrapolate the total allele copy number. By employing this technique, researchers have harnessed an elementary, yet potent, methodology for the delineation of macro-scale Copy Number Variations (CNVs). More granular regions of the genome—those deemed paramount for specific investigations—are probed using ultra-dense probes, often residing merely a few hundred nucleotides apart. To maintain comprehensive genome coverage, the interspersed genomic segments are analyzed using equidistantly placed main-strand probes. A note of caution, however, lies in the technique's inherent shortcomings, including an elevated false-positive propensity, fluctuating sensitivity metrics, and suboptimal concordance when juxtaposed across diverse platforms.
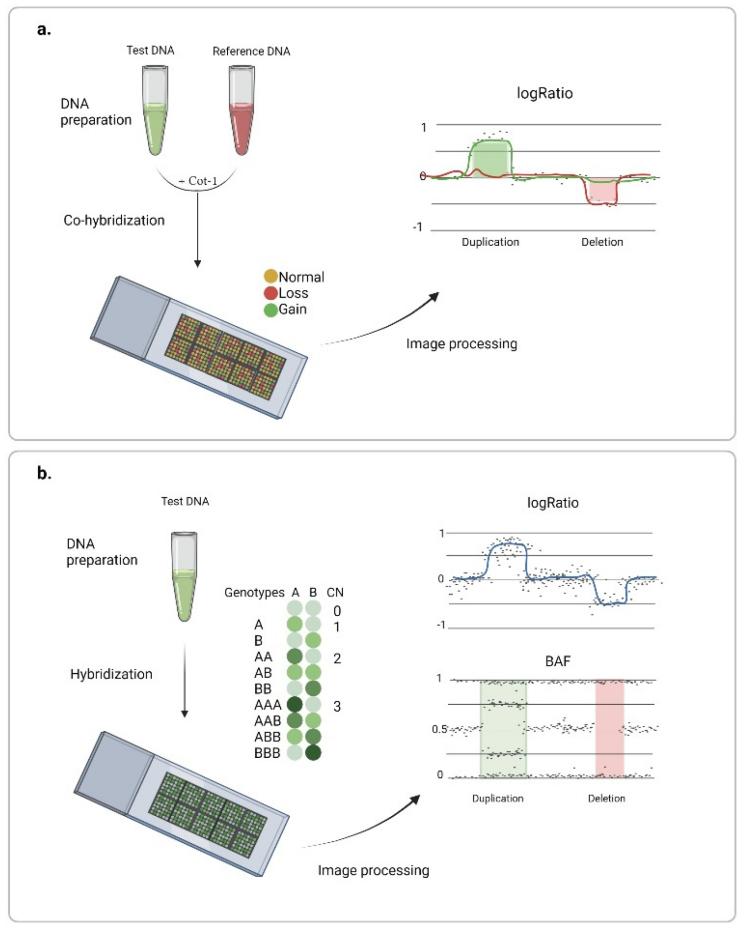
Chromosome microarray analysis. (Gordeeva V et al., 2022)
Concomitant with the ascendancy of high-throughput sequencing, especially next-generation sequencing (NGS) paradigms, CNV detection methodologies have undergone significant metamorphosis. This avant-garde technology employs a multifaceted approach towards CNV determination, encompassing strategies like read depth analysis, evaluation of inconsistent read pairs, exploration of split reads, and de novo assembly. The quintessence of short-read sequencing is its capacity for a refined numerical quantification of copy numbers, paired with an enhanced resolution capacity for minute variables (often less than 1 kilobase in size). Unlike its predecessors, this technique remains unencumbered by probe design limitations or inherent biases. Yet, the diminutive length of the reads combined with the labyrinthine nature of variant content invariably introduces hurdles in CNV delineation, thus leading to disparate caller performance metrics.
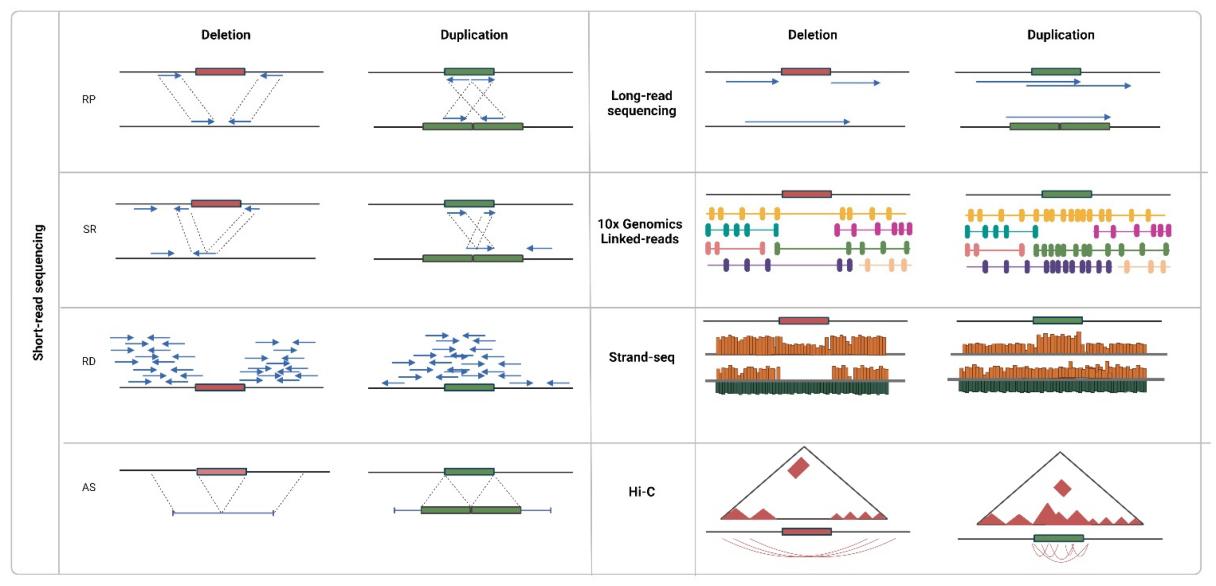
Approaches to CNV detection using sequencing data. (Gordeeva V et al., 2022)
PacBio HiFi sequencing proffers unparalleled advantages by furnishing single base pair resolution coupled with expansive reads—spanning multiple kilobases—encompassing a gamut of variants across the entire length. An attribute that renders this technology especially salient for CNV detection is the circumvention of sequence amplification, thereby drastically attenuating sequence coverage biases. Its most distinguishable merit, however, is its propensity to span extended repeat regions, thus furnishing an exhaustive genomic panorama and augmenting the probability of ensnaring substantial CNVs.
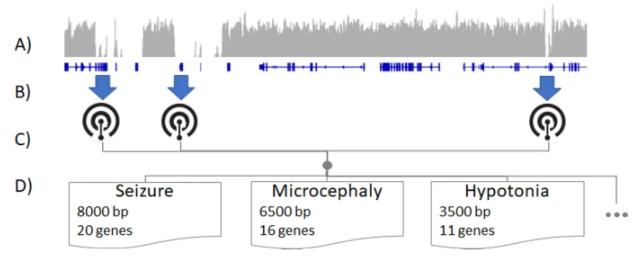
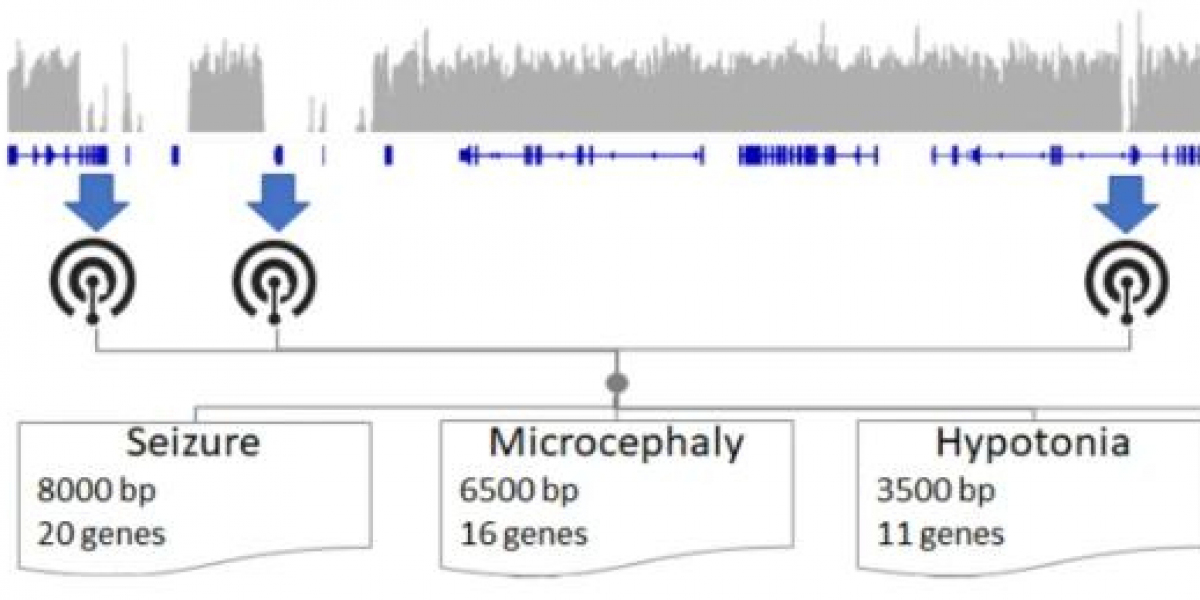
Illustrative schematic of determining HPO terms best assayable by LRS. (Sanford Kobayashi E et al., 2022)
HiFiCNV: A New Software Tool for CNV Analysis
The advent of HiFiCNV represents a transformative stride in CNV analysis. It not only facilitates the facile identification of CNVs from HiFi datasets but also permits meticulous visual scrutiny as necessitated. When synergistically utilized with variant datasets engendered by adjunct HiFi tools, researchers are equipped with an all-encompassing depiction of every variant archetype within a specimen. This equips them with a potent toolset, significantly amplifying the precision in rare disease analyses. Features intrinsic to HiFiCNV include:
- Optimized segmentation and calling
HiFiCNV is tailored primarily for whole genome sequencing (WGS) of HiFi germplasm, providing segmentation and calling capabilities that are distinct from traditional tools.
- GC bias estimation and correction
One of the outstanding features of HiFiCNV is its ability to automatically estimate and correct GC bias in sequencing data. This feature ensures that the integrity of the sequencing data is maintained, thus ensuring more accurate CNV predictions.
- Visualization and output format
HiFiCNV not only detects CNVs, it also visualizes them. By generating big-picture traces of capture depth and allele frequencies, researchers can visualize large-scale CNV events using the Integrated Genome Viewer (IGV) for deeper, more intuitive analysis. In addition, the versatility of the output formats, including bedplot and VCF, ensures compatibility with a wide range of downstream analyses.
- Efficiency and performance
HiFiCNV is equipped with multi-threading to ensure fast processing of sequencing data without compromising accuracy. When benchmarked against known CNVs, HiFiCNV demonstrated excellent capabilities in CNV detection, validating its position as the top tool for CNV analysis using PacBio HiFi reads.
References
- Gordeeva, Veronika, Elena Sharova, and Georgij Arapidi. "Progress in methods for copy number variation profiling." International journal of molecular sciences 23.4 (2022): 2143.
- Sanford Kobayashi, Erica, et al. "Approaches to long-read sequencing in a clinical setting to improve diagnostic rate." Scientific Reports 12.1 (2022): 16945.
- Lavrichenko, Ksenia, Stefan Johansson, and Inge Jonassen. "Comprehensive characterization of copy number variation (CNV) called from array, long-and short-read data." BMC genomics 22.1 (2021): 1-15.








